I got diagnosed with multiple sclerosis (again)
eestikeelne versioon
As I'm writing this I'm currently 11 days post-IVMP — which is basically three days of corticosteroids shot directly into my veins.
My right hand is just not currently working (but fortunately since a few days ago I started to be able to move my fingers, so it seems like hopefully everything is going to heal)
However, Multiple Sclerosis (MS) is a lifelong autoimmune disease.
We understand how it works — your immune system attacks the myelin sheath around your nerves (the insulation, basically) and you get lesions in your brain and spinal cord and things stop working properly — but medicine hasn't figured out the precise triggering mechanism. Why this person, why this moment.
There are clues — EBV infection, vitamin D deficiency, specific genes, latitude, gut health — and the research is closing in on EBV as likely necessary but not sufficient. But there's no complete answer yet.
The conventional way this is treated is with disease-modifying therapies (DMTs) — drugs that modulate or suppress parts of your immune system to slow the disease. Some are blunt immunosuppressants, some are more targeted (depleting specific B cells, blocking immune cell trafficking). They work for a lot of people. But they manage the disease — they don't address root cause.
And for me, that was never going to be enough.
I'm not a person to follow conventional by any means.
The first diagnosis 10 years ago changed me as a person — I can pinpoint it to that moment — and I don't take things lying down. I genuinely thought the first diagnosis was a misdiagnosis — right up until this happened.
Anyhow, here's my unconventional way of dealing with this...
I've divided this whole thing into parts - just jump to whatever you're interested in.
- The history — my first diagnosis, what it did to me as a person, and how everything I've built since ultimately traces back to this disease
- The relapse — what happened in March 2026 after a decade of nothing
- Conventional vs. integrative — my experience with both systems and what each one does and doesn't do
- What the system found — the tool I built, the findings, the genetics, the things no single doctor caught
- What I'm doing about it — the treatment plan, sequencing, and why order matters
- What I think I learned — reflections
The history
I'm not going to unpack the full history here — it would take way too long and honestly I've already written about it. I actually wrote about it in my first book. So instead of retelling the whole thing, here's the actual chapter.
Open the box below if you want the full story of how I got diagnosed the first time and everything that led up to it.
Read the full chapter from Mastering Fasting (my book, published in 2021)
(If you want the full book it's on Amazon Kindle. I also have a box of paperbacks sitting in my garage but I can't be bothered to figure out how to sell them right now.)
The short version: I was 24, living in Amsterdam when the symptoms started in December 2015.
I went home to Estonia for Christmas, a doctor flagged the symptoms, and the workup — MRIs, lumbar punctures, the whole thing — led to a formal MS diagnosis in early 2016 at ITK in Tallinn.
But here's the thing that made this confusing for a decade: alongside the MS diagnosis, I was also diagnosed with Lyme disease At the exact same time, two diagnoses at once...
And Lyme can cause demyelinating lesions that look like MS on an MRI.
So for years I genuinely believed the whole thing was a Lyme misdiagnosis — that the real problem was Lyme, not MS, and that once the Lyme was dealt with I was fine.
On top of that, I never actually had an episode. No symptoms, no attack, nothing. I just ended up being diagnosed through a chain of referrals — a doctor's visit led to a neurologist, which led to an MRI, which showed lesions. It was entirely accidental. When you've never actually felt anything wrong with you, it's easy to convince yourself they got it wrong. Turns out they didn't — but honestly, everything I did during those 10 years of "denial" is what got me to where I am now.
Back then, the neurologist told me to prepare for life-altering changes. The options at the time were self-injections every other day (interferon, glatiramer) with significant side effects for a ~30% reduction in relapse rate. Not nothing, but not exactly inspiring either. I refused all disease-modifying therapies.
However the landscape has changed since then — there are genuinely effective targeted therapies now — but my approach hasn't, because I was never interested in suppressing my immune system. I wanted to understand why it went wrong in the first place, and fix the underlying cause.
Knowing everything I know today, I'm still making the same call. Either the best or the worst decision of my life and I'm still not sure which one it is.
After refusing treatment I just... went deep researching this disease. Only problem was — I was a film major.
I had zero background in health, biology, any of this. I didn't understand what I was looking at. I was just scared and driven by denial and started reading everything I could find — disconnected things, no idea where to start, trying to piece something together from scratch.
So over the next 10 years I tried everything. All sorts of experimental and alternative treatments. I got into biohacking really early — before it was trendy, before the social media wellness crowd took it over. I changed my entire lifestyle, my diet, my habits. I got pretty deep into it. Deep enough that Estonian media started calling me a biohacker and I ended up writing a book about fasting. But honestly, after a while I got tired of it. The biohacking community became too hypochondriac, too performative. Social media overtook it. I stepped back from a lot of the health optimization stuff.
But looking back I imagine that to some extent that was actually to my own detriment — because a lot of what I was doing during those years was likely what was keeping the disease under control.
For 10 years after diagnosis I had zero clinical relapses, I lived normally without medication (if you discount injecting gray market peptides for fun).
When I wrote that book in 2020, I genuinely believed I had beaten it.
Then March 2026 happened — and everything I thought I knew turned out to be wrong.
The relapse
A month before the relapse, the whole family got a really bad stomach bug & I was throwing up for a full day & it got so bad that I had to go get an IV on day two because I couldn't stand up properly.
I recovered, and within a few days I was already back in the gym & doing two trainings a day - BJJ at 7 a.m. (which meant forcing myself awake at 6, something I hadn't been doing in a long time), then straight to the gym after. On off days I was doing infrared saunas and cold plunges.
I was also working really long days building Baby Acrobatics.
Then two weeks before the relapse my daughter was born (my second child) & month before that we'd moved to a new apartment because of a dispute with our landlord (Because the previous house had so many problems and uncontrollable dust issues which I wasn't having) and a month before that we moved back from Portugal to Estonia.
So it was stressor on stressor on stressor on stressor.
And my body just had enough.
March 31. I went to the ER in the morning thinking it was a spinal thing — maybe a disc, maybe something got caught wrong on a plane. Then a neurologist showed up, sat me down, and started talking in that tone. I knew what was coming the second I heard his voice & saw his mannerisms.
(the MS talk, again)
I walked out of there, took a methylcobalamin shot in my shoulder and wrote my friend Justin.
(Quick side note on Justin — Justin Maguire is a friend of mine in South Africa, deep into functional medicine. You give him your blood work and he just knows what's going on. Pulls together how everything connects — hormones, immune system, gut, methylation, all of it. We'd been working together for over a year at this point. If one person can figure this out, it's this dude.)
And honestly what I noticed is that the first thought that popped into my head wasn't about me — it was about my son. How am I going to pick him up? What do you tell a kid? "Daddy has to go to the doctor again"? "Sorry daddy can't walk anymore his legs don't work"??
April 1. Day two. Losing dexterity in the right hand. Left leg numb. Justin called — he's got my back, he's in my corner. He told me to drop cold plunges, cryo, infrared, everything immediately. Turns out the cold plunges and cryotherapy I'd been doing were likely making things worse — putting additional immune stress on an already overloaded system.
I was trying to be healthy and was actively doing damage to myself. MRI scheduled for Monday.
April 4. Day five. Symptoms progressing. My micro motor skills were getting worse — opening and closing my hand as fast as I could was... not fast. Then the Lhermitte's sign showed up: put my chin to my chest and I'd get electrical jolts shooting through my right hand. That's a demyelination signal — when the nerve is stripped of its insulation and bending the neck stretches it and triggers the current.
So new symptoms were appearing and everything was getting worse.
Meanwhile the costs were piling up...
- ER visit: €20
- Neurologist: €100
- Blood draws (58 markers): €970
- Glutathione + vitamin C IV: €300
- MRI with contrast: €420
- Supplements: €500+
AND I couldn't work properly.
Also these expenses were only the beginning — these numbers would quadruple within weeks.
So, yeah. It's not affordable being sick.
Then there was the waiting....
I needed to run a few more markers to check if this was viral reactivation (EBV, herpes viruses) — because if it was, I'd need antivirals immediately to stop progression, and steroids would actually make it worse. There's research showing that corticosteroids directly reactivate EBV at the molecular level — they literally tell the virus to wake up. And with my genetics (I'll get into this later but I have three genes involved in virus detection that are all compromised), that's not a theoretical risk.
But it was Friday. Everything was closed. Long weekend. Germany had holidays. Nothing moving.
For my personality, that's literally insufferable, especially since my symptoms are worsening.
April 7. Monday morning, 6 or 7 a.m. I woke up with this gut feeling that things were getting serious.
I just felt something changing, so I just drove to the ER of the hospital with the MS ward first thing.
They took me in, did tests, confirmed it — this is MS, active, happening right now. I saw the neurologist, did a quick detour to get my contrast MRI at a private clinic, came back, and they started me on intravenous methylprednisolone. Three-day course, ambulatory — I didn't have to stay overnight, just showed up each morning for the drip.
After the first day, temperature sensation started coming back on my left side. Positive sign. By the end of the three days I could flex my toes more, fingers started moving better. But my right hand was getting stiffer at the same time — stiff and tingling like crazy, like a dead appendix hanging off my body, throwing my balance off, causing my back to hurt when I walked. Getting better and feeling worse at the same time.
April 8. Day two of IVMP. The MRI results came in — and they found more than just the active C4 cervical lesion. Existing lesions in my brain that hadn't been there on previous scans. And T1 black holes — axonal damage. Permanent. Irreversible. The disease had been accumulating damage silently for 10 years while I thought I'd beaten it.
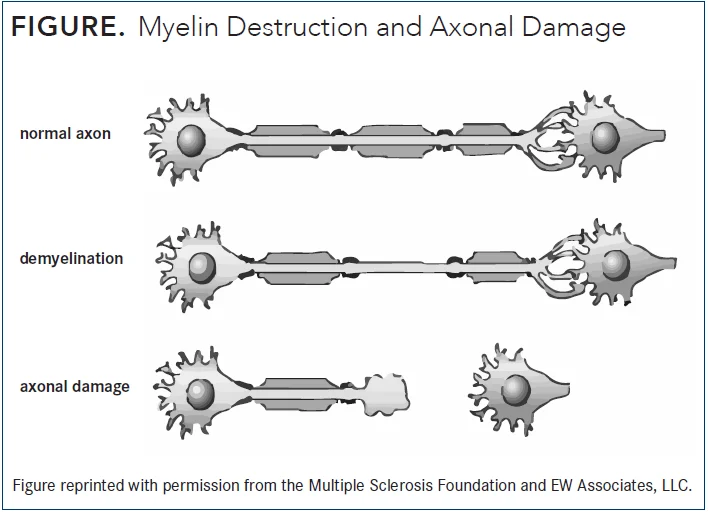
Your nerves are like electrical wires. Myelin is the insulation around the wire. In MS, the immune system strips the insulation off — that's demyelination. The wire (axon) is still there, and the body can partially rebuild the insulation. That's why symptoms often improve after a relapse.
But when the damage goes deeper and the wire itself gets destroyed — that's axonal damage. The wire is gone. That's what T1 black holes on an MRI show. That damage is permanent.
My MRI showed both: active demyelination (the current relapse, potentially repairable) and old black holes (accumulated over 10 years, not repairable).
So yeah, it's real. And now I had to actually deal with it.
That same day, sitting there with an IV in my arm and MRI results on my phone, I started building. I channeled my frustration and my fear into the only thing I know how to do — I opened my laptop and started building a tool to make sense of all of this. That's where everything you see below came from.
Conventional vs. integrative
When you have a disease like this you end up navigating two completely different medical worlds that don't talk to each other.
For most of the past 10 years I was defiant about this — maybe naive. It wasn't top of mind every day and honestly I forgot about it to an extent. The relapse showed me I shouldn't have.
That said, the diagnosis is the reason I am who I am.
It's why I started researching health, why I changed my lifestyle, why everything I've built since exists — Baby Acrobatics, all of it. If I hadn't been diagnosed, none of this would exist.
I'm still figuring out where the line is between integrative and conventional.
But I'm approaching it with a lot more respect for both sides than I had at 24.
The conventional approach
The standard treatment for MS is disease-modifying therapies — DMTs.
Some suppress your immune system broadly, some are more targeted (depleting specific B cells, blocking immune cell trafficking into the brain). They reduce relapses by 50-70% in trials.
For most people with MS, they're the right call.
I declined them all after my first diagnosis. I'm still declining them now.
The disease was clearly doing damage I didn't know about, so I'm not going to say my approach was perfect. But when this relapse hit, I didn't start looking at which DMT to go on. I started looking at what actually went wrong — at a molecular level, what broke, what caused this.
I'd rather fix the thing that's actually broken than suppress the immune system that's reacting to it.
But here's what the conventional system doesn't do: it doesn't look past the surface. The labs they run are vanity metrics — they confirm something exists but they don't tell you why. And the reference ranges they use are built from the average of whoever walks into a lab, which is an increasingly unhealthy population. So "normal" just means "not obviously broken." It doesn't mean healthy.
My TSH was 0.41. Reference range: 0.40 to 4.00. That's a hundred-fold span called "normal." Conventional doctors often don't even run the deeper thyroid markers — and when I ordered reverse T3 through Synlab myself, the lab technician told me it was the first time in years of working there that she'd seen anyone order that test. That's how rarely anyone looks at this stuff. Justin ran FT3, reverse T3, found my thyroid was functionally suppressed, and it changed the entire treatment approach.
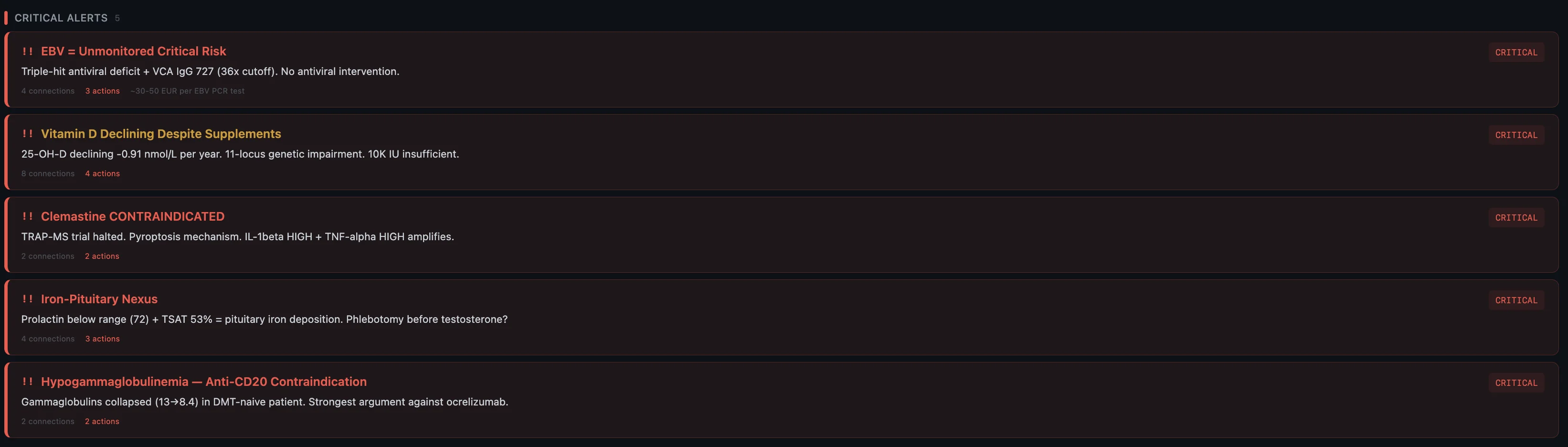
Or take the DMT they'd most likely want to put me on — fingolimod is one of the most commonly prescribed ones, and my system flagged that it's contraindicated with my melanoma history from age 20. A connection sitting in my medical history the whole time.
A concrete example: during my IVMP treatment I asked the hospital to run an EBV test. Of course they couldn't on a whim so I had to pay extra for it, fair enough.
I needed to know if the virus was reactivating, because corticosteroids can literally trigger EBV replication at the molecular level, and with my genetics that's a real risk. They said they'd do it.
It took them nine days. NINE days later I got the results — and they were qualitative, not quantitative. The difference is that qualitative just tells you yes or no, do you have the antibodies.
Quantitative tells you how much — the actual levels. I needed the levels.
What I got back was just "yes you have EBV antibodies." I've known that for years. That tells me nothing. Five or six calls to the hospital lab later, I found out they don't do quantitative EBV tests at all. Nobody mentioned this. I went to Synlab, paid another €100+ out of pocket, got the proper test, and found out my EBV antibodies are 35 times above the cutoff — persistent viral load that my genetics make nearly impossible to clear on my own.
So now I'm adding antiviral peptides to the protocol to deal with it. But I could have known this on day one. They also never tested MOG antibodies — if those came back positive, this wouldn't be MS at all but a completely different disease.
Don't get me wrong — I'm genuinely thankful they put out the fire. Western biomedicine is incredible at that, they have frameworks for it. But diagnostics and putting out fires is all they're good for (in my humblest of opinions).
The integrative approach
I actually found Justin because of mold, not MS.
Back in 2021 I moved to Portugal and within eight days of moving into a new house I got severe mold toxicity — black mold hidden in the walls. I was losing my eyesight, couldn't think, couldn't function.
Conventional doctors (I did see multiple specialists) had no idea what was going on. I needed someone who could actually read my labs and figure out what was happening inside my body, and that's when I found Justin.
He had me run an ungodly amount of blood work plus some other panels (OATs), looked at all of them, figured it out, put me on a protocol, and got me back on my feet. He also suspected that the mold exposure was connected to my MS — that I'd likely already had mold toxicity earlier in life and it may have contributed to the onset. Looking back, I think the Portugal mold episode is also when I had one of those silent MS episodes, because I was experiencing neurological symptoms at the time too.
That's the kind of thing integrative medicine does that conventional medicine doesn't.
Justin and his team at Autonomic Wellness handle the hormonal optimization, the supplement protocols, the methylation support, the stuff that conventional neurology doesn't touch.
When you actually look at the genetics and the bloodwork together — not just "is this value in range" but "why is this value where it is and what else does it connect to" — you start seeing things nobody was looking for.
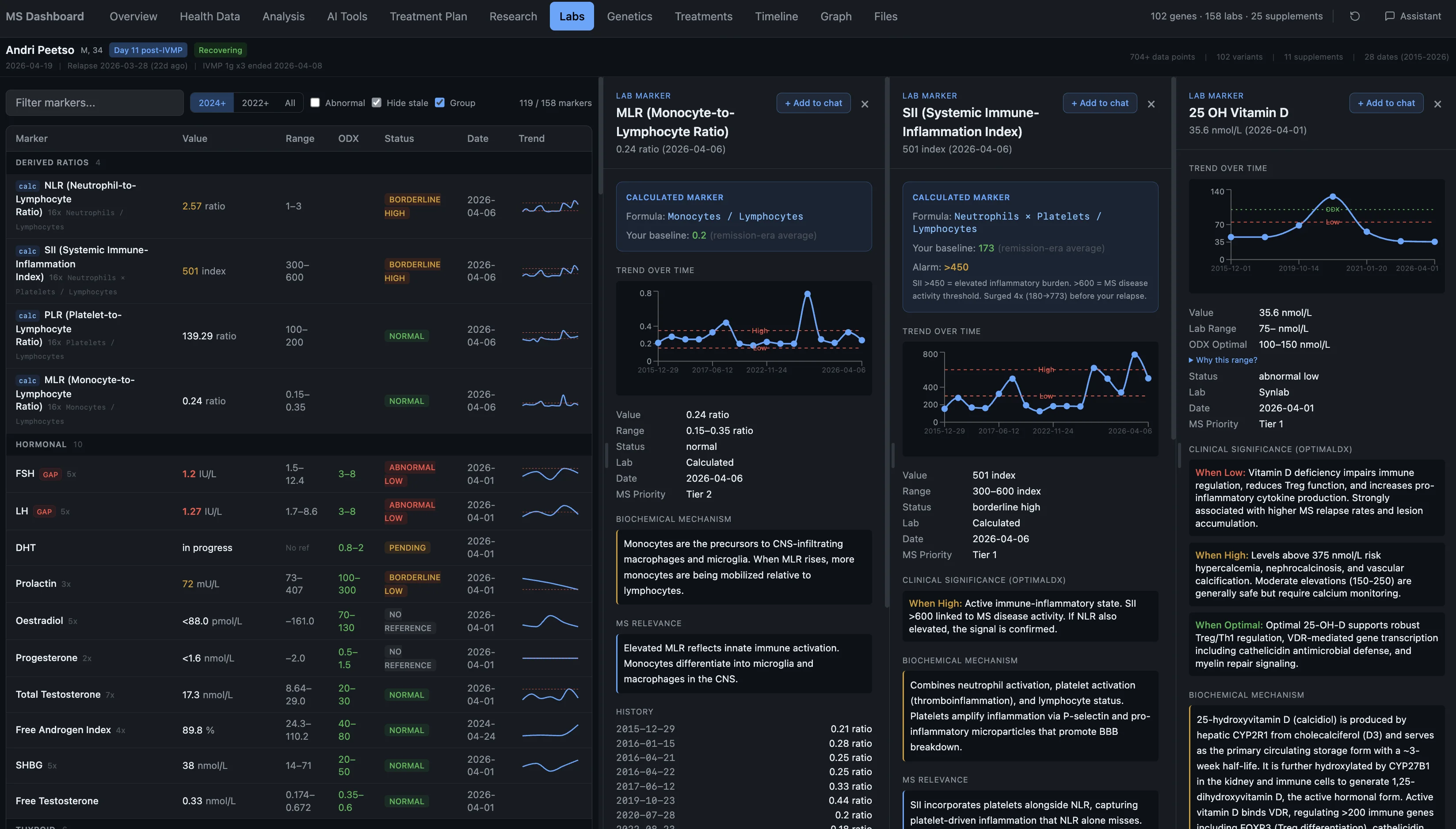
Take vitamin D. Conventional sufficiency is 75 nmol/L — anything above that and you're "fine." Functional optimal for someone with MS and my genetics is 100 to 150 nmol/L. Mine was 35.6. Not even close to either standard. And nobody in conventional medicine was going to tell me that 75 isn't good enough.
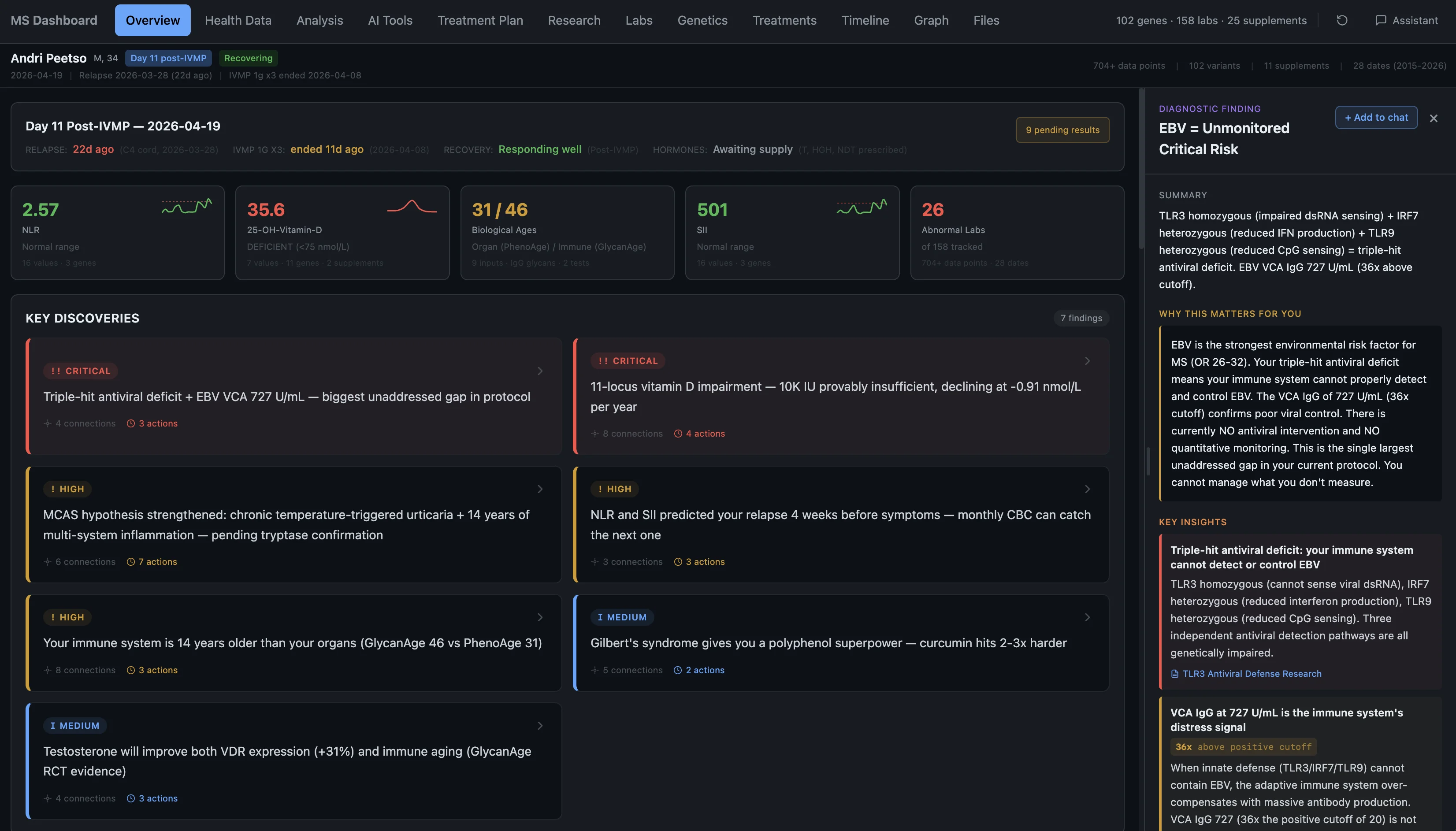
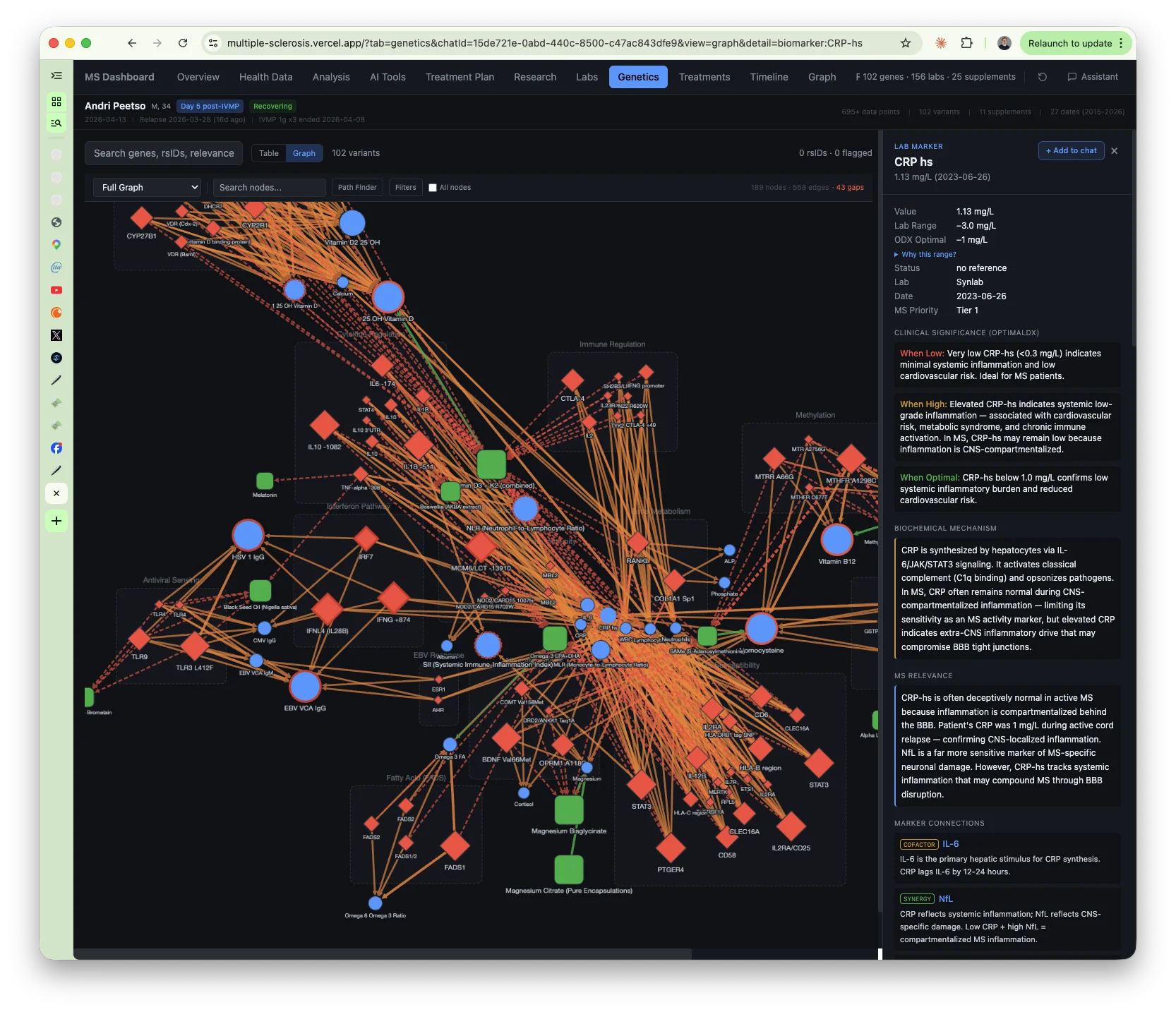
What the system found
I built a tool for myself that pulls together all my lab work, genetics, MRIs, supplements, and research into one place where everything cross-references everything else. It has an AI research layer, a knowledge graph that maps how genes connect to biomarkers connect to supplements, and an analysis engine that walks through pathways looking for patterns.
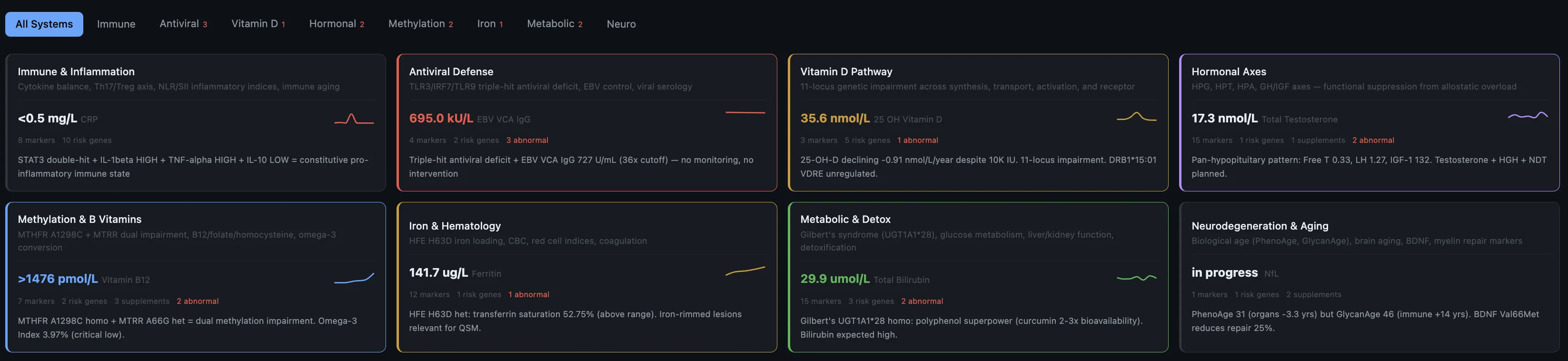
Before I get into the individual findings — here's the short version of what the system pieced together about why this happened now, after 10 years of nothing:
- Genetic predisposition — HLA-DRB1*15:01 (the strongest MS risk gene), homozygous hits on multiple inflammatory pathways, dual methylation impairment. The foundation was always there.
- Impaired virus control — triple-hit across TLR3, IRF7, and TLR9. Three genes involved in detecting and fighting viruses, all compromised. Combined with sky-high EBV that my body can't clear.
- Vitamin D pathway broken end-to-end — 11 variants across every step from synthesis to receptor. Not just "low vitamin D" — the entire metabolic chain is compromised.
- Thyroid conversion failure — DIO2 homozygous, can't convert T4 to T3 properly. T3 is what your brain needs to rebuild myelin after damage. So the repair mechanism itself is impaired.
- Mast cell hyperreactivity — 7 conditions across 14 years, all connected to mast cell dysregulation. The system tying inflammation, immune activation, and tissue damage together.
- Accumulated stressors — the stomach bug, the moves, the newborn, the training load, the sleep deprivation. One of them tipped the scale.
Each of these is a finding on its own. Together they tell a story.
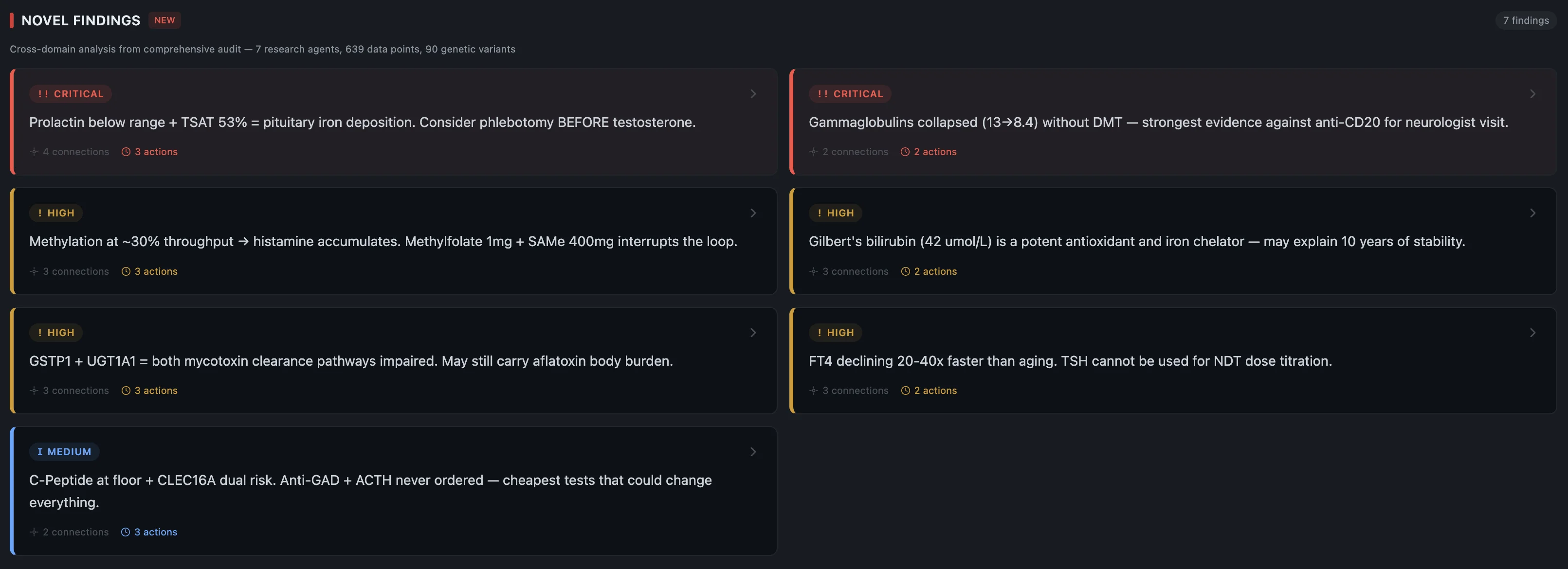
-
Multiple systems redlining — my genetics had me running on thin margins across virus control, vitamin D, thyroid conversion, mast cell regulation, and methylation. All compromised, all at once. Not one smoking gun — five of them.
-
The stomach bug — February 2026, severe enough to need an IV. This was likely the trigger. A massive immune activation event on a system already running at its limit.
-
Compounding stressors — newborn, house move, country move, training twice a day, waking at 6am, working long hours. Each one manageable alone. Together they were pulling resources from an immune system that had none to spare.
-
Cold exposure backfired — I was doing cold plunges and cryo regularly, thinking I was helping recovery. Instead I was adding immune stress on an already overloaded system. Actively doing damage to myself while trying to be healthy.
-
No monitoring — I had no system tracking my labs over time. If the NLR/SII spike pattern holds, a routine blood test during the stomach bug could have flagged something weeks before symptoms appeared.
-
The cascade — it doesn't take much when you're already at the edge. One more insult, one more stressor, and the whole thing tips. The stomach bug wasn't special — it was just the last thing.
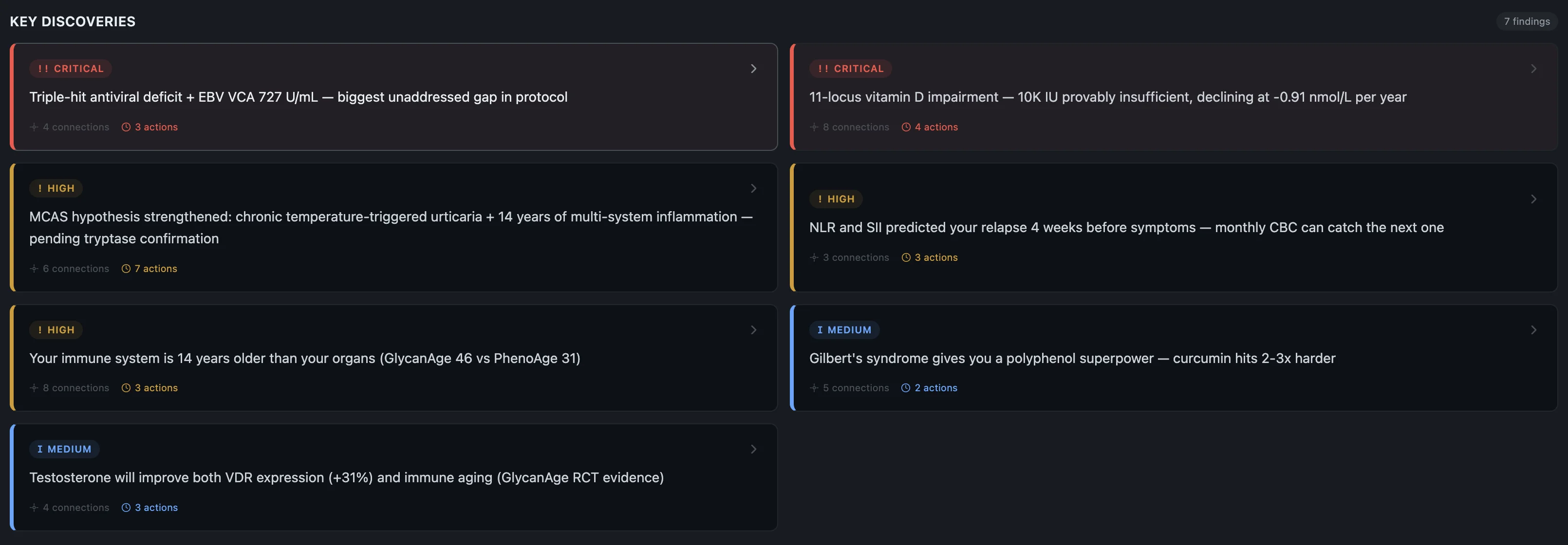
Here are the biggest findings:
The genetics
I extracted ~90 MS-relevant variants from my Estonian Biobank genome data and organized them into 14 categories.
MS susceptibility: HLA-DRB1*15:01 (odds ratio ~3.0, the strongest known genetic risk factor for MS), homozygous on two STAT3 variants (double-hit on the Th17 inflammatory pathway), IL2RA/CD25 homozygous risk, PTGER4 homozygous, RGS1 homozygous. Lots of homozygous risk alleles.
Cytokine imbalance: IL-1beta homozygous HIGH, TNF-alpha heterozygous HIGH, IL-10 homozygous LOW. In plain language my genetics produce a lot of pro-inflammatory signals and very little counter-regulation. Pro-inflammatory state with no brake.
More genetics: methylation, neuroplasticity, iron, thyroid, pharmacogenomics
Methylation: MTHFR A1298C homozygous + MTRR A66G heterozygous. Dual methylation impairment affecting folate metabolism, homocysteine clearance, histamine breakdown.
Neuroplasticity: BDNF Val66Met heterozygous, ~25% reduced BDNF secretion which impairs remyelination. Kind of important when your disease literally destroys myelin and you need your brain to rebuild it.
Iron metabolism: HFE H63D heterozygous, explains my elevated transferrin saturation (52.75%). Would be alarming without the genetic context — with it, it's expected and monitored.
Gilbert's syndrome: UGT1A1*28 homozygous. My bilirubin runs high (42 µmol/L) and it looks scary on a lab report but it's genetically benign. The dashboard knows this and overrides the severity from CRITICAL to LOW with documented rationale.
Thyroid: DIO2 Thr92Ala homozygous. Makes me a poor converter of T4 to T3 (the active thyroid hormone). Discovered during pharmacogenomics analysis, not something anyone was looking for. Turned out to be critical for treatment sequencing.
Pharmacogenomics: CYP3A5, CYP450 variants, UGT polymorphisms, OPRM1. These determine how I metabolize specific drugs and the system checks every new medication against this profile before it enters the treatment plan.
Good news: APOE ε3/ε3 (no Alzheimer's risk), no Factor V Leiden, no Prothrombin mutation, TPMT wild type.
The antiviral triple-hit
I have a triple-hit across TLR3-L412F (homozygous — impaired dsRNA sensing), IRF7 (heterozygous), and TLR9 (heterozygous). Three genes involved in detecting and fighting viruses, all compromised. Combined with sky-high EBV antibodies (VCA 727 U/mL) and the growing evidence that EBV is causally linked to MS — that pattern tells a story.
There's one compensating factor: IFNL4 TT gives me strong interferon-lambda production which partially makes up for the sensing deficit. But "partially" is doing a lot of work in that sentence.
And then COVID happened.
My pre-infection IgG was 0.7. Post-infection: 2,748 (not a typo). When your virus-sensing apparatus is genetically impaired, a massive immune activation event like that is not ideal.
The vitamin D 11-locus chain
I have 11 variants across the entire vitamin D pathway. Not one or two SNPs — eleven, spanning every single step from sun hitting your skin to the vitamin D receptor actually doing its job in the cell.
DHCR7 (synthesis), CYP2R1 (first hydroxylation — two variants), GC/VDBP (transport — three variants), CYP27B1 (activation — three variants), VDR (receptor — four variants), CYP24A1 (catabolism — two variants). Every. Single. Step.
The analysis engine found this — it walks every step of a metabolic pathway and checks for variants at each point. Vitamin D had hits at all of them.
This explains why my vitamin D declined from 68 to 36 nmol/L over 7 years despite supplementing and despite living in southern Europe for part of that time.
I wasn't just "not getting enough sun" — the entire metabolic pathway is compromised.
MCAS — the 14-year pattern
I have a 14-year multi-organ pattern: melanoma (in situ) at 20 (2012), chronic tonsillitis (2012), MS diagnosis (2016), IBS-D (2019), hidradenitis suppurativa (2020), seborrheic dermatitis (2024), chronic urticaria (2026).
Seven seemingly unrelated conditions across different organ systems seen by different specialists over more than a decade.
This is where the AI research swarm earned its keep. The dashboard has 10 specialized agents — each pulls a different angle (genetics, labs, literature) and they converge. The MCAS hypothesis came up across all of them: every condition on that list has documented mast cell infiltration in the literature, and 7 genetic pillars converge on mast cell hyperreactivity.
No single doctor saw this pattern. The neurologist manages MS. The dermatologist treated the HS and sebderm. The gastroenterologist diagnosed IBS-D. The allergist saw the hives. Each one only sees their organ system.
One hard rule with the AI: it's a research collaborator, never an authority. Justin's prescriptions come first, then the treating neurologist, then published evidence, then AI research.
I still trust Justin more than my AI.
The T3-histamine-rT3 vicious cycle
Justin identified this one and the AI assistant helped model the interaction loop: thyroid hormone (T3) exacerbates histamine release, inflammation increases, reverse T3 goes up, T3 effectiveness decreases, the thyroid intervention defeats itself.
Then the system flagged that I'm DIO2 Thr92Ala homozygous — a poor converter of T4 to T3.
Nobody was looking for this. It came out of the pharmacogenomics analysis as a side finding and turned out to change everything about the thyroid approach. And not just the thyroid approach — this connects directly to MS itself.
T3 is what tells your brain to rebuild myelin. That's its job — it's the signal that makes precursor cells mature into the cells that actually wrap your nerves with new insulation.
DIO2 is what makes T3 locally in the brain. Mine's broken. So the repair signal never arrives at the place where it's needed most. The repair mechanism itself is genetically impaired. Nobody was ever going to find this by looking at thyroid labs alone.
The conclusion: mast cell stabilization (ketotifen) must come BEFORE thyroid hormone.
If you start thyroid first, the T3 triggers histamine, the histamine triggers inflammation, the inflammation converts T3 to reverse T3, and you're right back where you started but now you're also itchy.
Treatment sequencing matters as much as the treatment itself.
Deep dive: the hypothyroid-remyelination connection
This is one of the most important findings for my specific case and I think it's underappreciated in MS in general. MS patients have a 2.3x higher rate of hypothyroidism than the general population. They share autoimmune pathways and genetic susceptibility. About 25% of MS patients develop a second autoimmune condition and thyroid disease is the most common one. Recent Mendelian randomization studies suggest a causal relationship between TSH levels and MS development.
But the mechanistic piece is what matters here.
T3 is required for remyelination. Full stop.
Your brain has oligodendrocyte precursor cells (OPCs) sitting around demyelinated lesions. These OPCs are waiting for the signal to mature into myelinating oligodendrocytes — the cells that actually rebuild the myelin sheath. That signal is T3. T3 binds TRβ1 receptors on OPCs, activates the KLF9 gene, drives cell cycle exit, and the OPCs start differentiating into mature oligodendrocytes that can wrap axons with new myelin. Without adequate T3, OPCs just sit there. They never mature. Your lesions don't repair.
In animal models of MS, exogenous T3 restores remyelination. Synthetic TRβ agonists reproduce the effect in vitro. No human clinical trials yet but the preclinical data is overwhelming.
My specific situation makes this worse — three separate hits:
Hit 1: DIO2 Thr92Ala homozygous. DIO2 is the enzyme in astrocytes that converts T4 to T3 locally in the CNS. My homozygous variant reduces DIO2 enzymatic efficiency by roughly 20-40%. Even with normal serum T3, my brain and spinal cord are T3-deficient. OPCs at my C4 lesion site are getting less T3 than they need to differentiate. This is invisible on standard blood tests — serum FT3 doesn't reflect CNS levels.
Hit 2: Inflammation upregulates DIO3. Active MS lesions upregulate DIO3, which degrades T3. So at the exact site where I need T3 most — my active C4 cord lesion — inflammation is actively destroying it. Less T3 production from DIO2 being broken, plus more T3 destruction from DIO3 being upregulated, equals a remyelination block.
Hit 3: My serum thyroid is already suppressed. TSH was 0.41 (floor of range), FT3 was 4.08 pmol/L (sub-optimal, target is above 4.5). If my serum T3 is already sub-optimal, and my CNS T3 is further reduced by 20-40% from DIO2... I'm looking at critically low T3 at the lesion site.
And it's not just thyroid. All four pituitary axes are functionally suppressed — thyroid (TSH 0.41), gonadal (free testosterone 0.33, LH 1.27), growth (IGF-1 132), adrenal (cortisol 286 low-borderline). But thyroid is the one that directly controls remyelination.
Why this matters for recovery: my C4 lesion is fresh, only weeks old. OPCs are recruited to acute lesion sites. If I can get adequate T3 to those OPCs in the next 3-6 months — the remyelination window — there's a real shot at functional repair. That's why Justin's treatment sequencing is so critical: testosterone first (neuroprotective, reduces inflammation), then ketotifen to stabilize mast cells (4-8 weeks to full effect), then natural desiccated thyroid last — only when the mast cell terrain is controlled so T3 actually reaches receptors instead of being converted to reverse T3.
Without adequate CNS T3, no amount of CDP-choline, phosphatidylcholine, or B12 can drive OPCs to mature. The T3 story is the remyelination story.
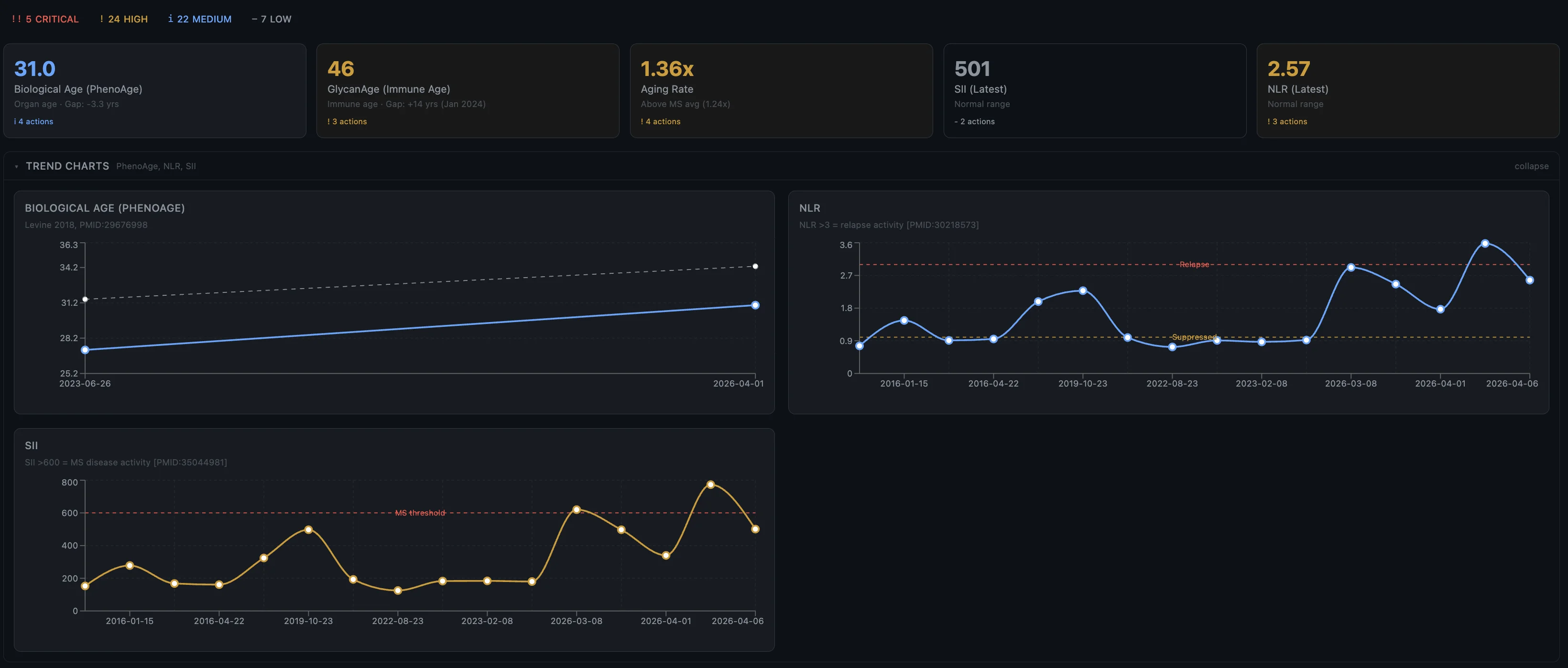
Biological age divergence
PhenoAge (biological age from 9 routine blood biomarkers): 3.3 years younger than chronological. The organs are in good shape.
GlycanAge (immune age from IgG glycosylation): +14 years. My immune system is aging 14 years faster than my organs.
That 17-year divergence is itself a finding. My immune system is chronically overactivated and aging faster than the body it's supposed to be protecting and is instead attacking.
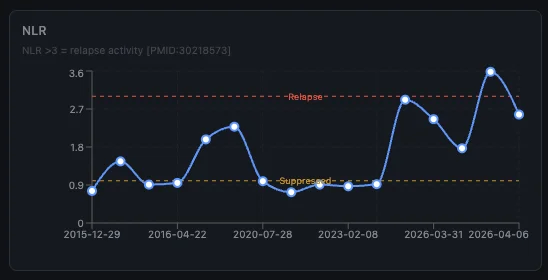
Inflammatory ratios as early warning
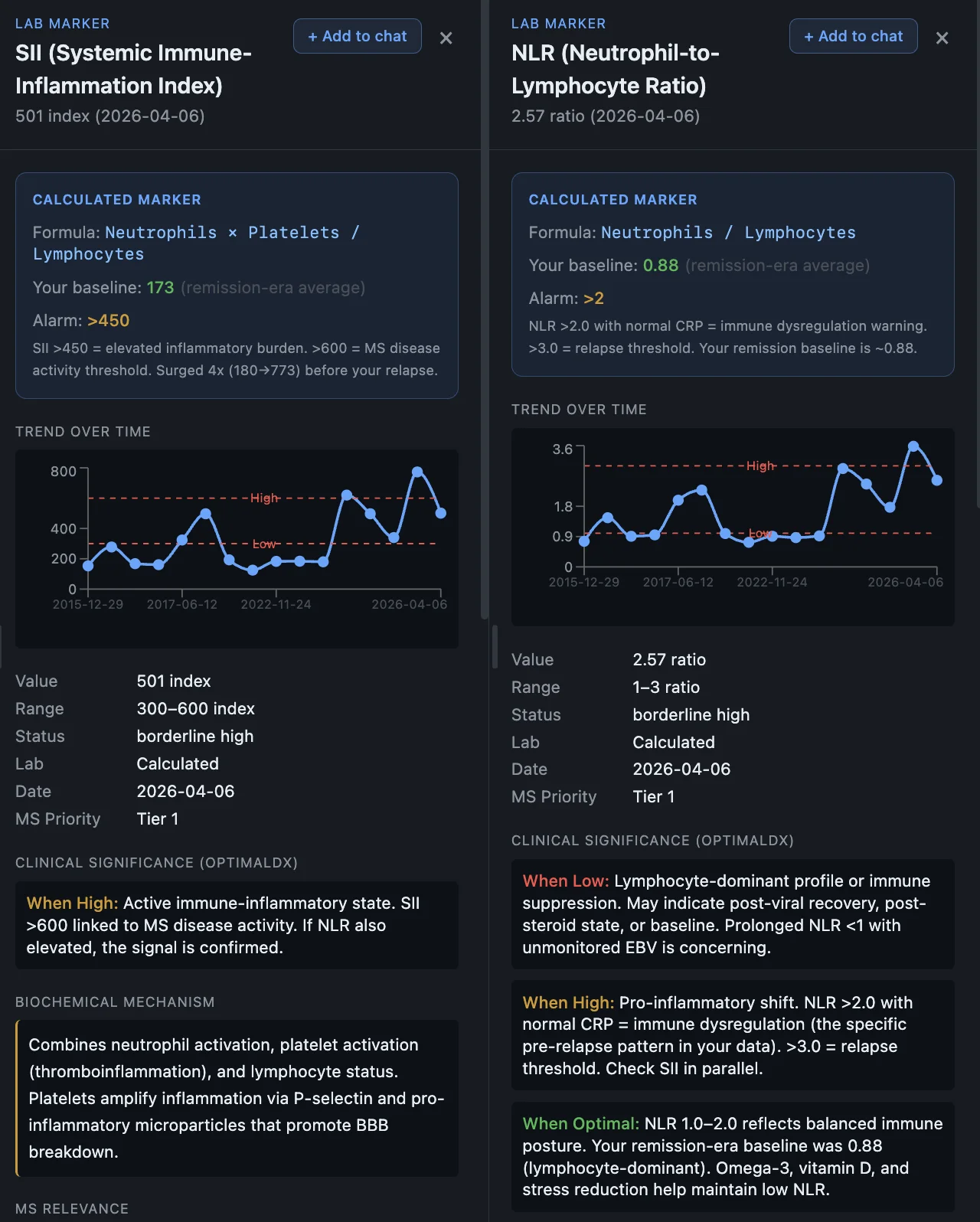
The dashboard tracks inflammatory ratios over time. Looking back, my NLR and SII spiked about 4 weeks before the March 2026 relapse. If that pattern holds it means a routine blood test could warn me before I even feel symptoms.
If I had this system tracking my labs when I went to get that IV during the stomach bug, I could have probably flagged something early.
What I'm doing about it
Findings are useless without action. Here's what I'm actually doing — every intervention has a genetic or lab-verified rationale, every sequencing decision has a mechanism. Nothing is random.
The core insight is that four hormonal axes are simultaneously suppressed — thyroid, testosterone, growth hormone, and adrenal — and all four need to be fixed, but in the right order. You can't just throw everything at the wall. The order matters because some interventions will sabotage others if you do them wrong.
-
Testosterone first — my free T is 0.33 nmol/L (crashed). Justin pushed for this immediately. Testosterone is neuroprotective independent of its anti-inflammatory effect — the only clinical trial of testosterone in MS men (Sicotte 2007, UCLA, n=10) showed a 67% reduction in brain atrophy rate and significant cognitive improvement. It also increases IL-10 production (my genetics make me a low IL-10 producer), shifts immune balance away from Th17 (which attacks myelin), enhances VDR expression by 31% (critical given my 11-locus vitamin D impairment), and expands CD8+ T cells for EBV surveillance.
-
Ketotifen (mast cell stabilizer) — already prescribed at 2mg daily. This has to be established before thyroid optimization because T3 triggers histamine release from mast cells. If mast cells aren't stabilized first, any T3 supplementation will trigger histamine → inflammation → reverse T3 → self-defeat. Takes 4-8 weeks to reach full effect.
-
Low-dose naltrexone — I was on LDN before the relapse. Once the acute phase settles, I'm going back on it. TLR4 antagonism on microglia, suppresses IL-1β, triggers endorphin upregulation.
-
Thyroid optimization — only after mast cells are stabilized. My DIO2 is broken so I can't convert T4 to T3 properly, and T3 is the master signal for remyelination. The pending reverse T3 result will determine how aggressive mast cell stabilization needs to be before this starts.
-
Growth hormone optimization — IGF-1 at 132 is at the floor. IGF-1 is non-redundant for OPC differentiation — knockout mice show near-total remyelination failure. Justin prescribed peptide support for this. Target IGF-1: 200-250 µg/L.
-
Antiviral peptides — Thymosin Alpha 1 + Hepatitis peptide for EBV control, given the triple antiviral defense impairment and EBV antibodies at 35x above cutoff.
Deep dive: the clinical evidence for testosterone in MS
There's exactly one clinical trial of testosterone treatment in men with MS — Sicotte et al. 2007, UCLA, published in Archives of Neurology (PMID:17708679). It's small (n=10) but the results are hard to ignore.
The setup: 10 men with RRMS, not on any DMTs, applied testosterone gel daily for 12 months. Each patient served as his own control — 6 months of just watching, then 12 months of treatment. Same delivery method as my planned Testogel. Their average total testosterone was 17.1 nmol/L — mine is 17.3. Essentially identical baseline.
What happened:
- Brain atrophy rate dropped from -0.81%/year to -0.26%/year — a 67% reduction in brain volume loss (P<.001). That's comparable to what high-efficacy DMTs achieve, except it's a hormone, not an immunosuppressant.
- Cognitive scores on the PASAT (working memory and processing speed) improved significantly at 12 months (P=.008).
- Lean muscle mass increased by 1.7kg.
- Zero adverse effects over 12 months.
The most interesting part: it was NOT anti-inflammatory. The number of enhancing lesions (active inflammation) didn't change. The brain protection happened without reducing inflammation. This means testosterone works as a direct neuroprotectant, not as an immunosuppressant. The likely mechanism is aromatization — the body converts some testosterone into estradiol, which activates ERβ receptors on oligodendrocyte precursor cells (the cells that make new myelin).
The second paper — Olsen & Kovacs 1995 (Am J Med Sci) — is a different disease (lupus) but the same principle. A man with Klinefelter's syndrome and severe lupus had his testosterone dose increased. Within 9 months, his autoantibodies dropped from 1:320 to 1:10 (essentially normalized), and his CD8+ T cells expanded from 17% to 22%. CD8 expansion is directly relevant to me — those are the cells responsible for keeping EBV in check, and my triple antiviral defense impairment (TLR3 + IRF7 + TLR9) means I rely heavily on CD8 cells to compensate.
Why my genetics predict a stronger response than average: my IL-10 is genetically suppressed (testosterone directly increases IL-10 production — fills a critical gap), my STAT3 double-hit skews me toward Th17 (testosterone counteracts this), my VDR has 4 variants (testosterone enhances VDR expression by 31%), and my BDNF is reduced (Val66Met — testosterone-driven BDNF boost matters more when you make less of it genetically).
Timeline expectation: neuroprotection manifests at 3-6 months, cognitive improvement at 9-12 months. This is a long-game intervention.
The remyelination window
My C4 lesion is fresh — weeks old. OPCs are recruited to acute lesion sites. The window for remyelination is roughly 3-6 months post-relapse, which is why the hormonal optimization timeline is so critical. Every week that passes without adequate T3 and IGF-1 at the lesion site is a week those OPCs aren't maturing into myelinating oligodendrocytes.
The four non-redundant remyelination pathways I need to fix simultaneously:
- Testosterone/androgen (free T 0.33) → restores androgen receptor-mediated OPC differentiation + enhances VDR
- Thyroid/T3 (FT3 4.08, target >5.0) → master OPC differentiation signal, but requires mast cell stabilization first
- GH/IGF-1 (IGF-1 132, target 200-250) → drives myelin gene expression (MBP, PLP, MAG, MOG) and myelin lipid synthesis
- Vitamin D/VDR (25-OH-D 35.6, target 125-175 nmol/L) → suppresses c-Myc OPC brake, controls HLA-DRB1*15:01
Testosterone + GH together enhance VDR expression by about 30%, which partially compensates for my 11-locus vitamin D impairment. This isn't additive — it's synergistic. All four axes need to be optimized together.
The three things that matter most right now: get my testosterone up, fix my thyroid, and get proper vitamin D levels. Everything else — ketotifen, LDN, antiviral peptides, the supplement stack — is supporting those three priorities. Right now my hormones are wrecked and that's what needs to change first. There's a lot of smaller optimization happening in parallel but the main levers are hormonal.
Why I'm not on DMTs
I know this is the part where most neurologists would disagree with me. Disease-modifying therapies are the standard of care for MS. I've declined them twice now — once at 24, and again at 34.
I don't trust them. Not because they don't work — they do reduce relapses. But they don't cure anything. They don't fix what's actually broken. They modulate your immune response so it attacks your myelin less frequently, but the underlying disease process continues. Your hormones stay crashed. Your vitamin D stays low. Your EBV stays uncontrolled. Your mast cells stay hyperactive. Everything else keeps decaying while the drug masks the symptoms.
The conventional approach is: take your DMT, keep living your life, come back for your MRI every year. Nobody looks at why your immune system went wrong in the first place. Nobody checks your testosterone or your thyroid or your antiviral genetics. The DMT is the treatment. That's it.
And the side effects aren't trivial. These are immunosuppressive drugs — they work by making your immune system weaker. For someone with my genetics, that's a serious problem.
| What my genetics say | What it means for DMTs |
|---|---|
| TLR3/IRF7/TLR9 triple-hit (impaired virus detection) | Every DMT that lowers immune surveillance = amplified infection risk. My body already can't detect viral replication properly. |
| IL-10 homozygous LOW (broken anti-inflammatory brake) | DMTs that cause immune reconstitution rely on IL-10 to prevent secondary autoimmunity — I can't make enough. |
| Melanoma at 20 | S1P modulators (fingolimod, siponimod, ozanimod) are contraindicated. These are the most commonly prescribed oral DMTs. Gone. |
| Gilbert's syndrome + HFE iron loading | Hepatotoxic DMTs (teriflunomide) stress a liver that's already handling excess iron and bilirubin. |
My melanoma history alone eliminates an entire class of DMTs. My antiviral genetics make natalizumab (the one with PML risk — a fatal brain infection from JC virus reactivation) particularly dangerous because my TLR3 literally can't detect the virus replicating. My IL-10 deficiency makes alemtuzumab's 40% secondary autoimmunity rate even worse for me than for most people.
Deep dive: DMT-by-DMT risk analysis for my profile
Interferons (Avonex, Rebif, Plegridy) — modest efficacy (~30% relapse reduction). Hepatotoxicity risk with my Gilbert's + HFE. Can trigger thyroid autoimmunity (5-10% of patients) and my thyroid axis is already fragile. My IFNL4 TT already gives me strong endogenous interferon-lambda — I'm partially doing what this drug does, naturally.
Glatiramer acetate (Copaxone) — safest DMT for my profile but weakest efficacy. Wouldn't have prevented my cord lesion. Post-injection reactions can mimic anaphylaxis — provocative for suspected MCAS.
Dimethyl fumarate (Tecfidera) — causes lymphopenia in 25% of patients. With my antiviral triple-hit, losing lymphocytes is catastrophic. PML risk (~1:3,000).
S1P modulators (fingolimod, siponimod, ozanimod) — contraindicated. Melanoma history. Full stop.
Natalizumab (Tysabri) — very effective but carries PML risk (4:1,000 if JCV positive). JC virus has dsRNA intermediates that my broken TLR3 can't detect. This is probably the most dangerous DMT for my specific genotype. Also causes devastating rebound MS when stopped.
Anti-CD20 (ocrelizumab, rituximab) — best mechanistic fit because it kills the B-cells making cross-reactive EBV antibodies. But it also kills B-cells making protective antibodies against everything else. With my antiviral deficit, I need every antibody I have. Melanoma signal in trials. B-cells take 6-12 months to come back after stopping.
Cladribine (Mavenclad) — skin cancer signal 3-5x. Lymphopenia for months. No.
Alemtuzumab (Lemtrada) — highest efficacy but 40% secondary thyroid autoimmunity. I already have a DIO2 double-hit and a fragile thyroid. My IL-10 low genotype means my immune system can't self-regulate during reconstitution — secondary autoimmunity risk is amplified.
The 10-year gap proves something. I went a full decade with no DMT and no clinical relapses. Yes, silent damage happened — the MRI shows that. But this relapse had clear precipitants — everything I described above hitting at once. This wasn't progressive disease grinding me down.
I'm not doing nothing. I'm addressing the root causes that DMTs don't touch — EBV persistence, hormonal collapse, mast cell dysregulation, vitamin D impairment, antiviral deficit. If the hormonal optimization and antiviral protocol don't stabilize things, I'll revisit. Stem cells are a possibility if I need to go further. Step by step.
Moving to the tropics
I'm looking for a location near the equator to move to with my family. This isn't lifestyle tourism — with 11 genetic variants across my entire vitamin D pathway, I need real sun exposure, not supplements alone. My vitamin D declined from 68 to 36 nmol/L over 7 years despite supplementing and despite living in southern Portugal. Estonia at 59°N latitude doesn't produce meaningful UVB for about 6 months of the year.
Thailand is the current frontrunner — good healthcare access, affordable, and my body might actually be able to produce vitamin D there.
Monitoring
Labs every 8 weeks for the first 6 months: full hormone panel (testosterone, estradiol, FT3, FT4, rT3, IGF-1, cortisol), inflammatory markers (NLR, SII, CRP), vitamin D with calcium and PTH. MRI at 3 months and 6 months. Neurofilament light chain as a biomarker for ongoing neuronal damage.
If the NLR/SII early warning pattern holds, a routine blood test could flag a relapse weeks before symptoms appear. That's the monitoring system I didn't have before.
What I think I learned
Structure beats volume. 640 lab data points are useless as a PDF stack. Put them in a CSV with canonical naming and alias resolution and cross-references and genetic context and they become something you can actually reason about.
Cross-referencing is where the value is. No single data source tells the story. The MCAS hypothesis only emerged when the system connected 7 conditions across 14 years with the genetic profile.
No individual specialist had that view.
AI agents need constraints more than they need capabilities. The Facts Before Theories rule, the authority hierarchy, the immutable sources policy — those constraints are what make the system trustworthy.
Nobody is going to connect your data for you. Your neurologist doesn't have time. Your GP doesn't have the context. And they're not gonna vibe code you a dashboard.
The only person with access to all the data and the means to analyze it is you.
I don't know if this is useful for anyone else. It's built for exactly one patient with exactly my genetic profile. But I think the idea is generalizable — treating your medical data as a knowledge graph where genes connect to biomarkers connect to supplements and everything is evidence-linked and AI-queryable.
Whether any of this actually changes outcomes or whether I just built an elaborate coping mechanism is honestly an open question.
The MCAS finding feels real. The NLR/SII relapse prediction feels real. The vitamin D 11-locus chain explains something that genuinely confused me for years. But I'm also the person who built the system so of course I think the findings are real.
"Healthy" practices can hurt you if you don't know what's actually going on inside your body. I was doing cold plunges, cryotherapy, training twice a day, waking up at 6am — convinced I was optimizing. In reality I was piling stressors onto an already compromised immune system. The cold exposure was adding immune stress. The overtraining was suppressing my hormones. The sleep deprivation was wrecking my recovery. I thought I was doing everything right and I was actively making things worse. You can biohack yourself into a relapse if you don't understand the terrain you're working with.
I'll probably keep building this thing for as long as I have MS, which is (presumably) forever.
As of writing this, my right hand is still not fully working but at least I'm moving again.
(My wife is not happy about any of this. Especially because instead of enjoying her post-pregnancy period and bonding with the baby, she suddenly had a husband who couldn't walk or take care of himself. And two screaming children.)